
FDA Clearance and the 510(k) Process for Medical Devices

Navigating the world of medical device regulation can be daunting, especially when it comes to understanding the role of the U.S. Food and Drug Administration (FDA) in ensuring the safety and effectiveness of medical products.
Two key terms you’ll often encounter are “FDA clearance” and “510(k) process.” Let’s break these down to understand their significance and how they impact the medical device industry.
What Is FDA Clearance?
FDA clearance, also referred to as 510(k) clearance, is a term used to indicate that the FDA has reviewed a medical device and determined it is safe and effective for its intended use. This process involves rigorous field and laboratory testing to ensure that a medical device, drug, or product is safe and effective for its intended use. Laboratory tests assess product safety, while field testing evaluates real-world performance, ensuring compliance with FDA standards before clearance for public use. This clearance is particularly important for medical devices that are classified under Class I or Class II. On the other hand, Class III devices go through a FDA approval process, called Premarket Approval (PMA). The distinction between FDA-approved and FDA-cleared refers to different regulatory pathways that medical products take to reach the market in the United States.
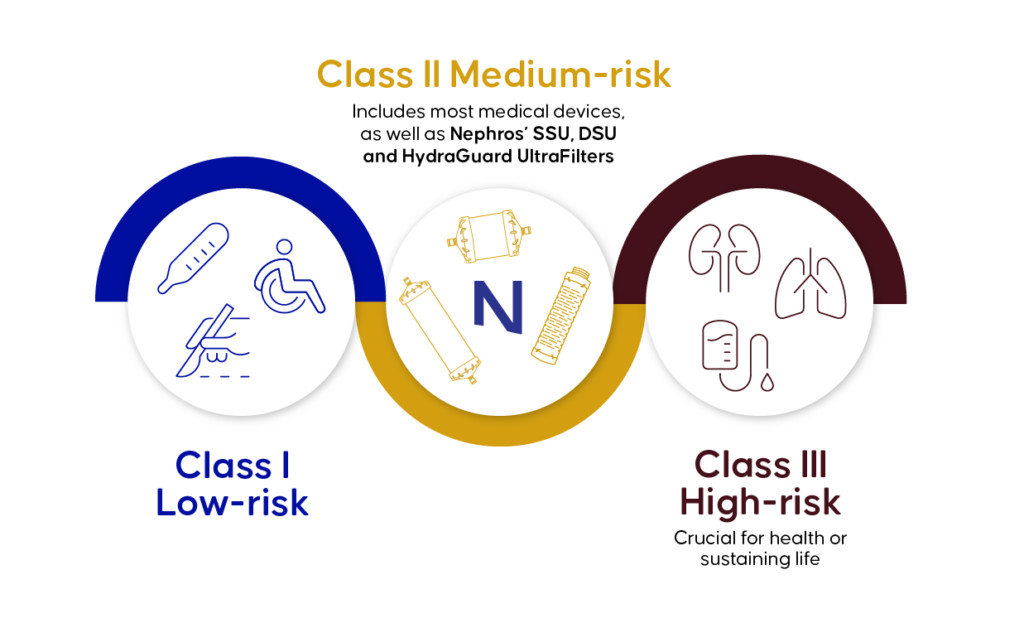
FDA Classification of Medical Devices
- Class I: Low-risk devices, such as bandages, handheld surgical instruments, or non-electric wheelchairs.
- Class II: Intermediate-risk devices, which include most medical devices, including Nephros infection control and dialysis filters.
- Class III: High-risk devices that are crucial for health or sustaining life such as, pace makers, heart valves, and cochlear implants.
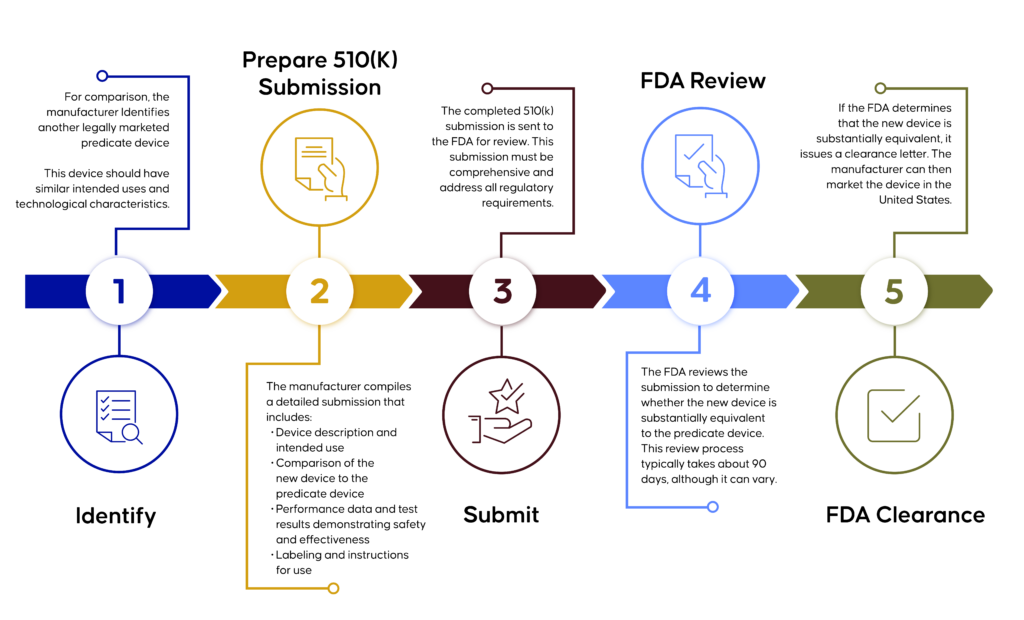
Overview of the FDA 510(k) Clearance Process
The FDA 510(k) process is a comprehensive, multi-step procedure that begins with identifying a predicate device and preparing a detailed submission. This submission is then reviewed by the FDA, a process that typically takes up to 90 days. Upon determining that the new device is substantially equivalent, the FDA issues a clearance letter, after which the manufacturer is authorized to market the device in the United States.
Post Clearance Responsibilities
Achieving FDA clearance is a significant milestone, but it’s not the end of the regulatory journey. Manufacturers must comply with ongoing FDA requirements, including:
- Good Manufacturing Practices (GMP): Ensuring the device is manufactured consistently and meets QMS standards, as well as 21 CFR 820 compliance.
- Post-Market Surveillance: Monitoring the device’s performance in the real world and reporting any adverse events to the FDA.
- Labeling and Promotion: Ensuring all marketing materials are truthful and not misleading.
How to Check FDA Clearance
To check FDA clearance, you can look up the drug or biological product on the FDA’s website. Most FDA-approved human drugs and therapeutic biological products allow you to search by drug name, active ingredient, or application number (NDA, ANDA, or BLA)
Vashone R. Thomas, Vice President, Quality & Regulatory at Nephros shares, “At Nephros, the FDA 510(k) clearance process is a key pillar in our commitment to delivering high-quality water filtration products that meet the stringent safety and efficacy standards required for healthcare and/or infection control settings. This clearance not only validates our technology but also reinforces our dedication to protecting patients and supporting infection control. By choosing Nephros, healthcare providers can trust that they are using reliable, FDA-cleared solutions designed to safeguard water quality. Our focus on innovation, compliance, and quality sets us apart as a leader in the industry.”